Reference



OpenAI's Answer
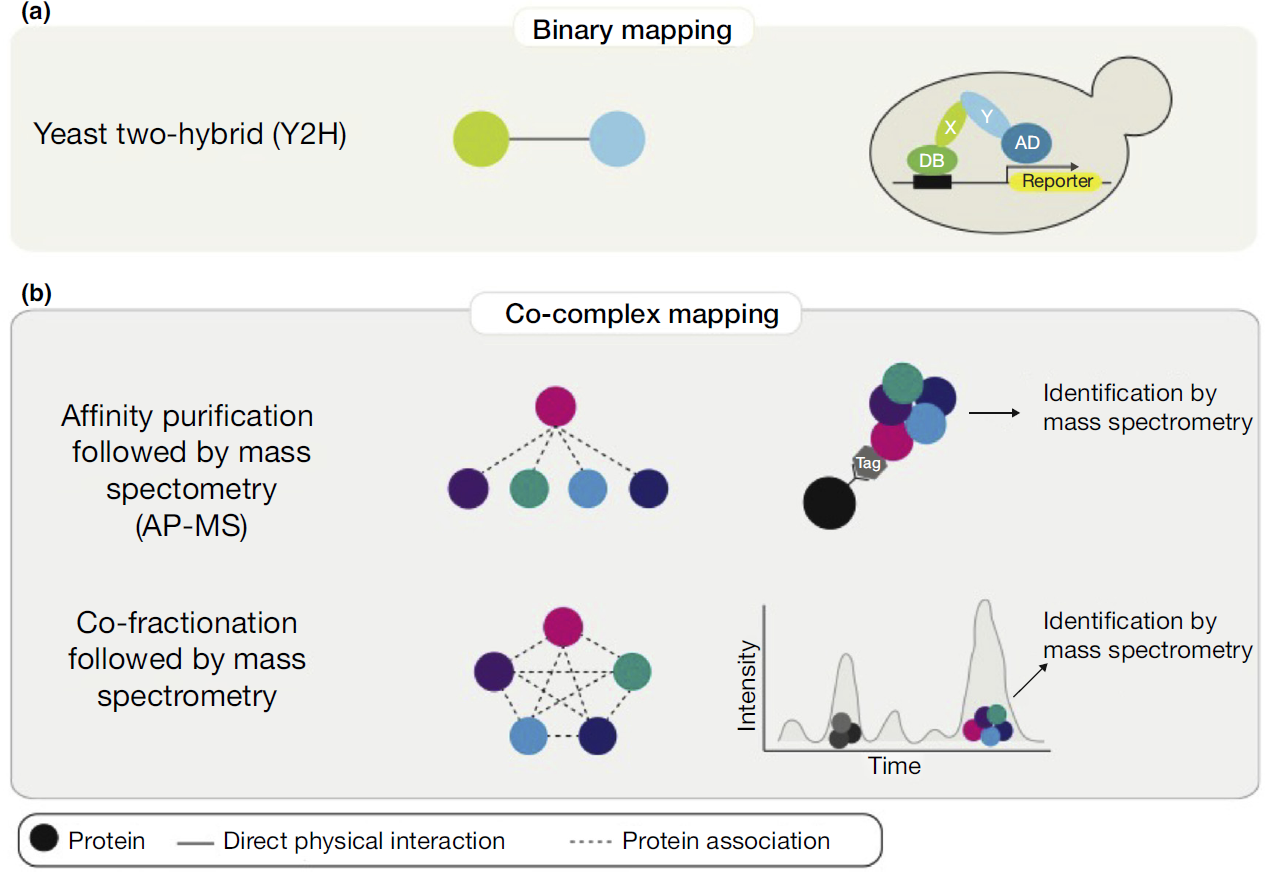
Large-scale mapping of human protein-protein interactions (PPIs) using mass spectrometry involves several advanced techniques. Here are some key methodologies used for this purpose:
-
Affinity Purification Mass Spectrometry (AP-MS):
- This technique involves the purification of protein complexes using specific antibodies followed by mass spectrometry to identify the components. It is widely used due to its ability to maintain the physiological context of PPIs.
- More about AP-MS can be found here.
-
Cross-linking Mass Spectrometry (XL-MS):
- XL-MS involves chemically cross-linking interacting proteins or protein regions in vivo, which can then be identified and analyzed using mass spectrometry. It is particularly useful for capturing transient or weak interactions.
- Learn more about XL-MS techniques.
-
Proximity-labeling Techniques:
- Methods like BioID and APEX use enzymes that label neighboring proteins within a certain radius, which can then be identified by mass spectrometry. These methods help in studying proteins that are in close proximity, possibly interacting within the cellular space.
-
Tandem Mass Tag (TMT) Labeling:
- TMT-based quantification allows multiplexed quantitative mass spectrometry, providing high throughput capability, essential for large-scale interactome studies.
-
Co-fractionation Mass Spectrometry:
- This involves fractionating cell lysates and analyzing the fractions with mass spectrometry. The co-migration of proteins across fractions can indicate protein interactions.
![Techniques for An-scale mapping of human protein–protein interactions (PPIs) by mass spectrometry involves several advanced techniques. Here are some prominent methodologies:
-
Affinity Purification Mass Spectrometry (AP-MS):
- AP-MS involves the isolation of protein complexes from a cellular extract through an affinity purification process. It is usually combined with mass spectrometry for the identification and quantification of interaction partners. This method is widely used for proteome-wide studies of PPIs.
- For additional details, refer to the study on AP-MS in F1000Research.
-
Cross-Linking Mass Spectrometry (XL-MS):
- XL-MS involves chemically cross-linking protein complexes, which stabilizes them and allows for the mapping of the spatial organization of proteins within complexes. This method provides insights into the structure and function of the interactions.
- More on this can be explored in the article from Nature Communications.
-
Immunoprecipitation Mass Spectrometry (IP-MS):
- Similar to AP-MS, IP-MS uses specific antibodies to enrich protein complexes before mass spectrometry analysis. This method helps in focusing on specific protein complexes and validating their interactions.
- An extensive discussion of the method is available on the EMBO Press website.
-
Multi-Dimensional Protein Identification Technology (MudPIT):
- MudPIT enhances protein identification by using multidimensional chromatography coupled with mass spectrometry. This technique allows for the analysis of complex protein mixtures and is suitable for large-scale interactome studies.
-
Protein Correlation Profiling (PCP-MS):
- PCP-MS involves the fractionation of cell extracts to separate protein complexes based on their size or other physicochemical properties, followed by mass spectrometric analysis. This method helps in determining co-purifying protein partners.
These techniques are crucial for dissecting complex biological processes and have been instrumental in advancing our understanding of the human proteome. Images and further insights can be found in Analytical Science Journals.
Each method provides unique insights and, when combined, delivers a comprehensive understanding of protein interaction networks and functionalities.
Follow Up
Related
What are the Catholic charities affiliated with the Archdiocese of New York?
How does Emma plan to use Mr. Gold to pull Excalibur from the stone?
How did Samuel Davis encourage the Daniell artists to explore the Himalayas?
What influence does Frank O'Hara's poetics have on Dmitry Kuzmin's poetry?
What role did Nicolas Tindal play in translating Paul de Rapin's History of England?
How many international caps did Patsy Gallacher earn for Ireland?